Fast Facts

Malignant PEComa is an ultra-rare sarcoma that appears to arise most commonly at visceral (especially gastrointestinal and uterine), retroperitoneal, and abdominopelvic sites
Globally, there is ≤ malignant PEComa diagnosis per 1,000,000 people annually, with an estimated 100-300 new patients per year in the US*
Patients are predominantly females and typically diagnosed between the ages of 45-65 years
Advanced malignant PEComa is associated with a poor prognosis
51% to 72% of patients with malignant PEComas develop metastatic disease within 12 to 23 months
*Aadi analysis based on multiple sources, including Aadi internal data and external research conducted by Tessellon Group and Corsica Life Sciences.
PEComa = Perivascular Epithelioid Cell Tumor
What is PEComa?
PEComas are a rare subset of soft tissue neoplasms composed of histologically and immunohistochemically distinctive epithelioid cells. Due to their rarity and relatively recent recognition, classification of PEComas has evolved; they may be broadly grouped into the following tumor types.
Malignant and Benign PEComa in Context
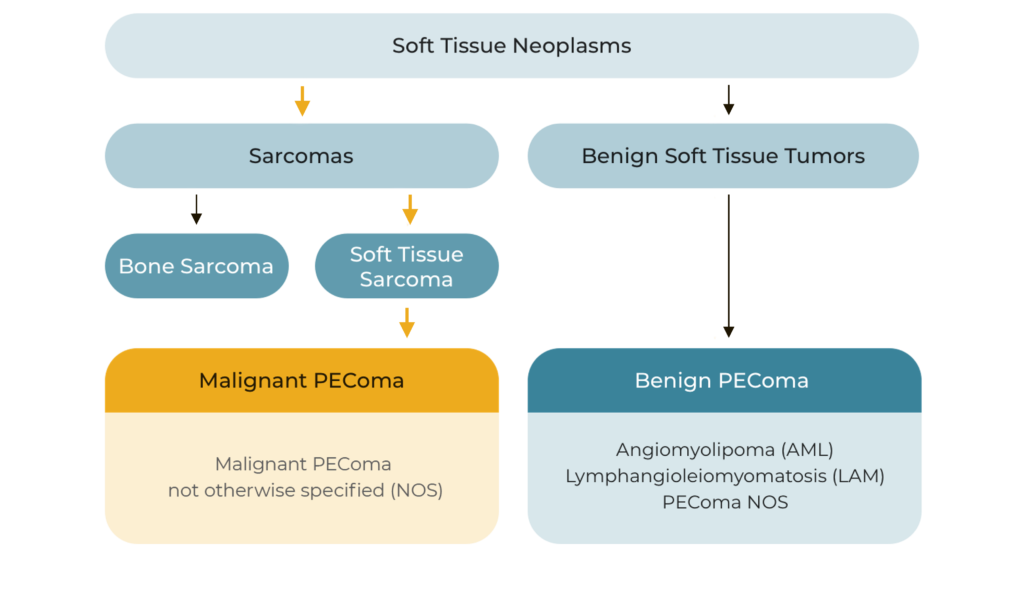
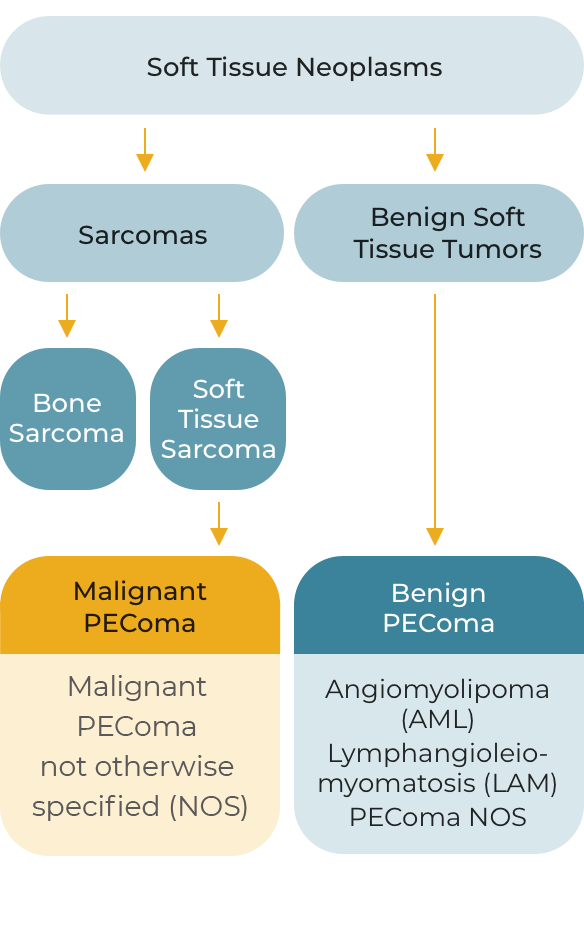
Although most PEComas are localized and benign, malignant PEComa is characterized by locally invasive recurrence or distant metastatic lesions. Approximately 72% of patients with malignant PEComa will develop metastatic disease, most frequently to the lung or liver, over a median follow-up of 12-months.
What does PEComa look like?
PEComas are characterized by the World Health Organization as mesenchymal tumors composed of distinctive perivascular epithelioid cells that show a focal association with blood vessel walls, and usually express both melanocytic and smooth muscle markers.
-
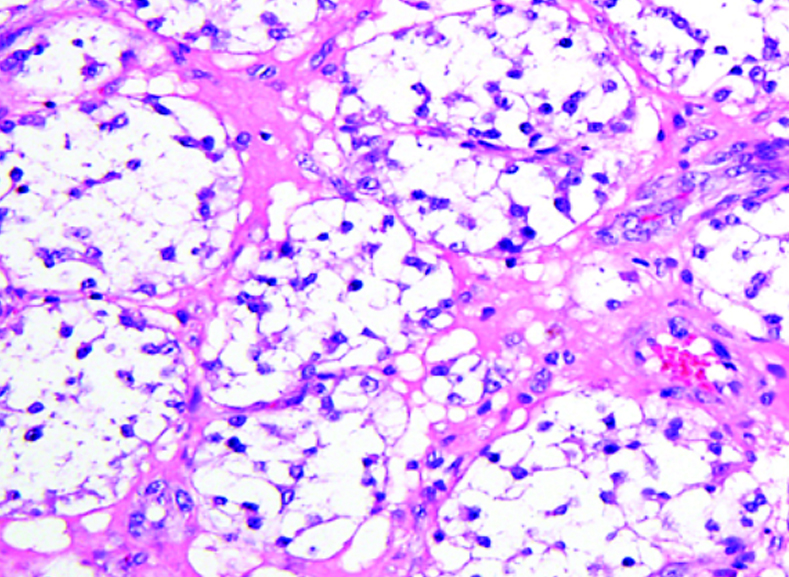
- Nested architecture and clear cell features
-
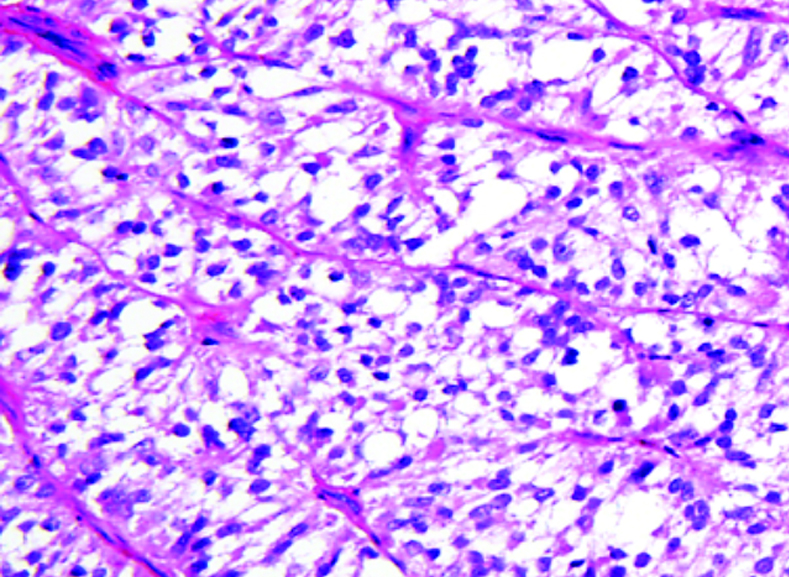
- Trabecular architecture and capillary vascular network
-
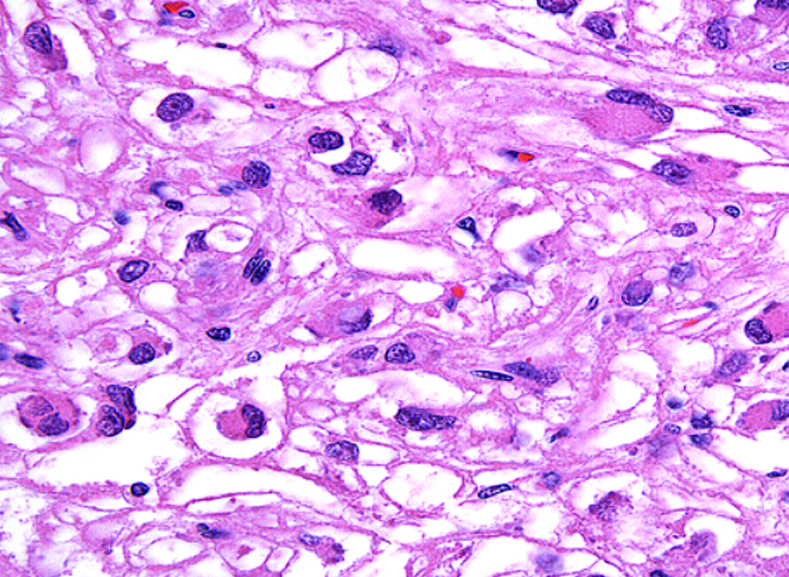
- Granular eosinophilic cytoplasm
-
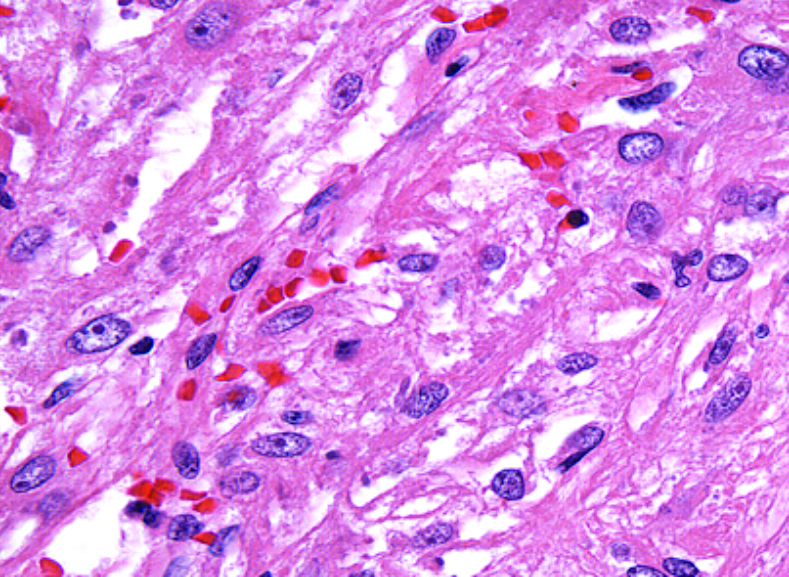
- Smooth muscle-like spindle cell morphology
Features of Malignancy
-
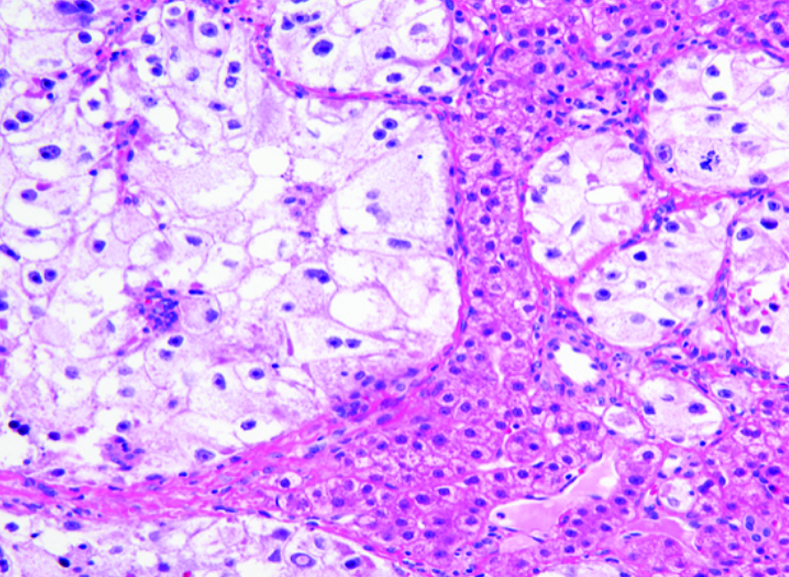
- Advanced malignant metastatic PEComa: nested architecture, clear cell morphology, and mitotic activity
-
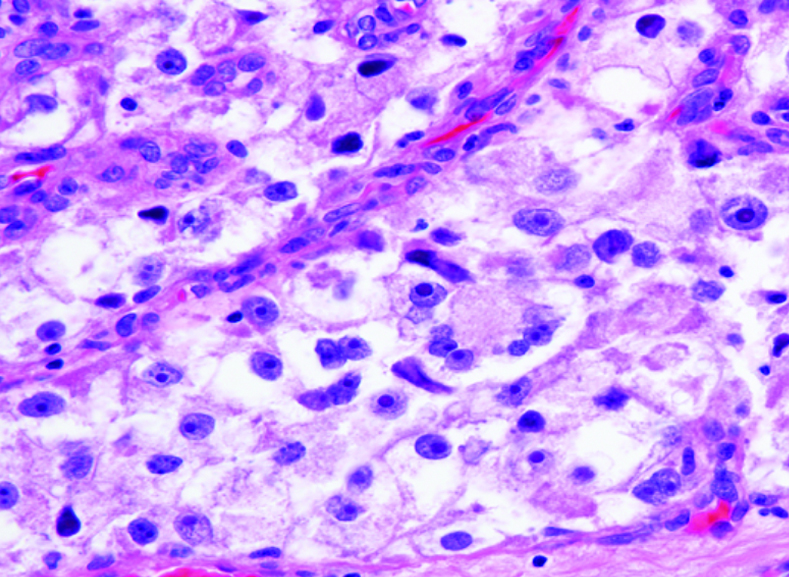
- Advanced malignant PEComa: marked nuclear atypia including macronucleoli and multinucleate cells
From the personal image library of Jason L Hornick, MD, PhD; 2020.
Diagnosis of advanced malignant PEComa is based on a pathological evaluation, though misdiagnosis may occur due to overlapping immunophenotype (characteristic co-expression of melanocytic and smooth muscle markers) and histologic overlap with other tumor types.
Potential misdiagnoses may include:
- Leiomyosarcoma
- Clear cell sarcoma
- Metastatic melanoma
- Gastrointestinal stromal tumor (GIST)
- Renal cell carcinoma (RCC)
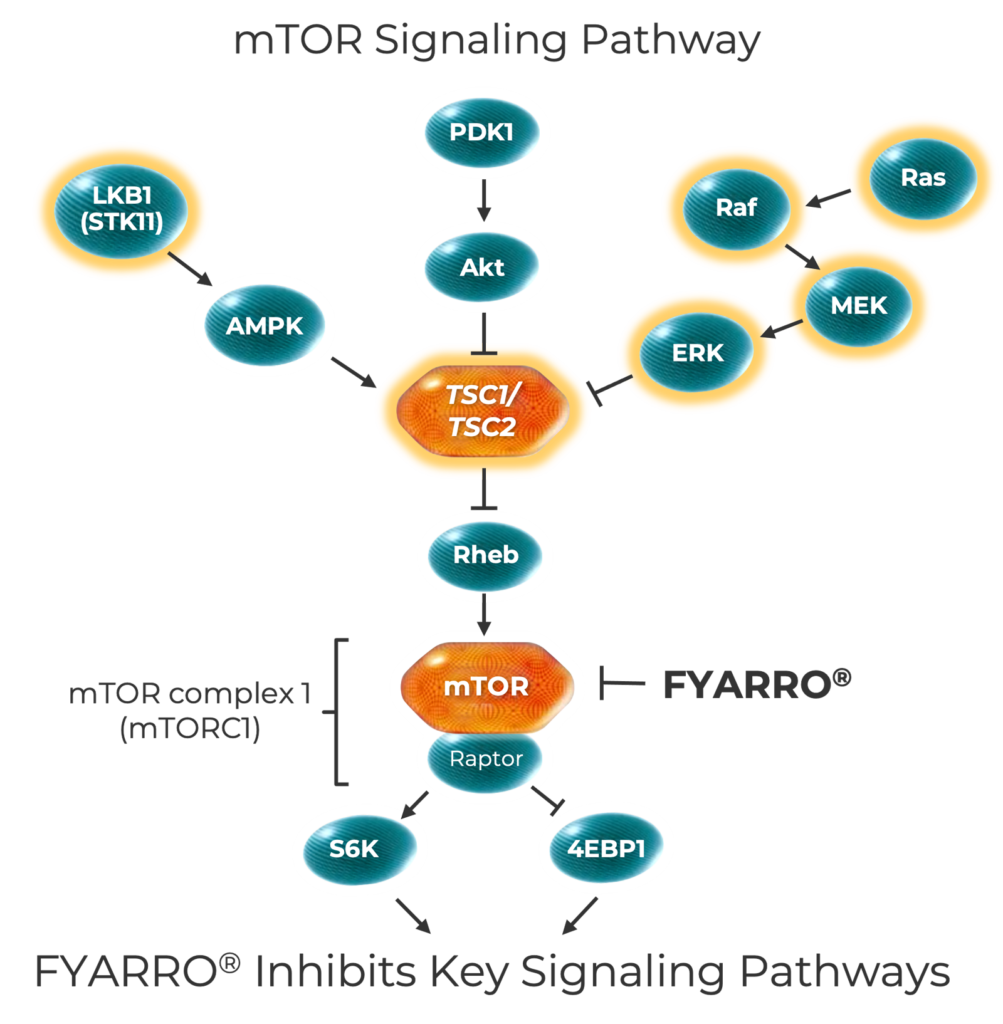
The mTOR Pathway and PEComa
The mTOR pathway controls key cellular processes. Alterations or deletions in mTOR pathway genes, like TSC1 and TSC2, may lead to mTOR overactivation, which prompts uncontrolled cell growth, metabolism, and survival. Advanced malignant PEComa tumors have the highest rate of TSC1 and TSC2 alterations among cancers.
mTOR=mechanistic target of rapamycin; TSC1=tuberous sclerosis-1; TSC2=tuberous sclerosis-2.